Machine learning to accelerate screening for Marcus reorganization energies
Omri D. Abarbanel, Geoffrey R. Hutchison. “Machine learning to accelerate screening for Marcus reorganization energies” J. Chem. Phys. 155, 054106 (2021). DOI Preprint Data
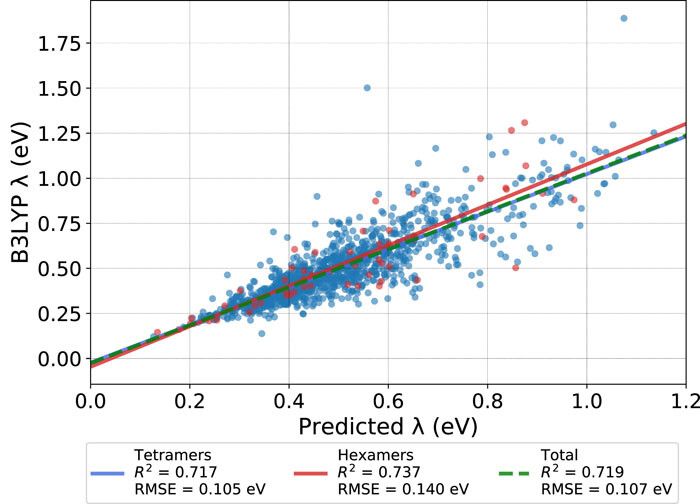 While many machine learning (ML) methods, particularly deep neural networks, have been trained for density functional and quantum chemical energies and properties, the vast majority of these methods focus on single-point energies. In principle, such ML methods, once trained, offer thermochemical accuracy on par with density functional and wave function methods but at speeds comparable to traditional force fields or approximate semiempirical methods. So far, most efforts have focused on optimized equilibrium single-point energies and properties. In this work, we evaluate the accuracy of several leading ML methods across a range of bond potential energy curves and torsional potentials. The methods were trained on the existing ANI-1 training set, calculated using the ωB97X/6-31G(d) single points at nonequilibrium geometries. We find that across a range of small molecules, several methods offer both qualitative accuracy (e.g., correct minima, both repulsive and attractive bond regions, anharmonic shape, and single minima) and quantitative accuracy in terms of the mean absolute percent error near the minima. At the moment, ANI-2x, FCHL, and a new libmolgrid-based convolutional neural net, the Colorful CNN, show good performance.
While many machine learning (ML) methods, particularly deep neural networks, have been trained for density functional and quantum chemical energies and properties, the vast majority of these methods focus on single-point energies. In principle, such ML methods, once trained, offer thermochemical accuracy on par with density functional and wave function methods but at speeds comparable to traditional force fields or approximate semiempirical methods. So far, most efforts have focused on optimized equilibrium single-point energies and properties. In this work, we evaluate the accuracy of several leading ML methods across a range of bond potential energy curves and torsional potentials. The methods were trained on the existing ANI-1 training set, calculated using the ωB97X/6-31G(d) single points at nonequilibrium geometries. We find that across a range of small molecules, several methods offer both qualitative accuracy (e.g., correct minima, both repulsive and attractive bond regions, anharmonic shape, and single minima) and quantitative accuracy in terms of the mean absolute percent error near the minima. At the moment, ANI-2x, FCHL, and a new libmolgrid-based convolutional neural net, the Colorful CNN, show good performance.