Deep learning coordinate-free quantum chemistry
Matthew K Matlock, Max Hoffman, Na Le Dang, Dakota L. Folmsbee, Luke A. Langkamp, Geoffrey R. Hutchison, Neeraj Kumar, and S. Joshua Swamidass.”Deep learning coordinate-free quantum chemistry” J. Phys. Chem. A 2021, 125, 40, 8978–8986 DOI {: .btn .btn–success}
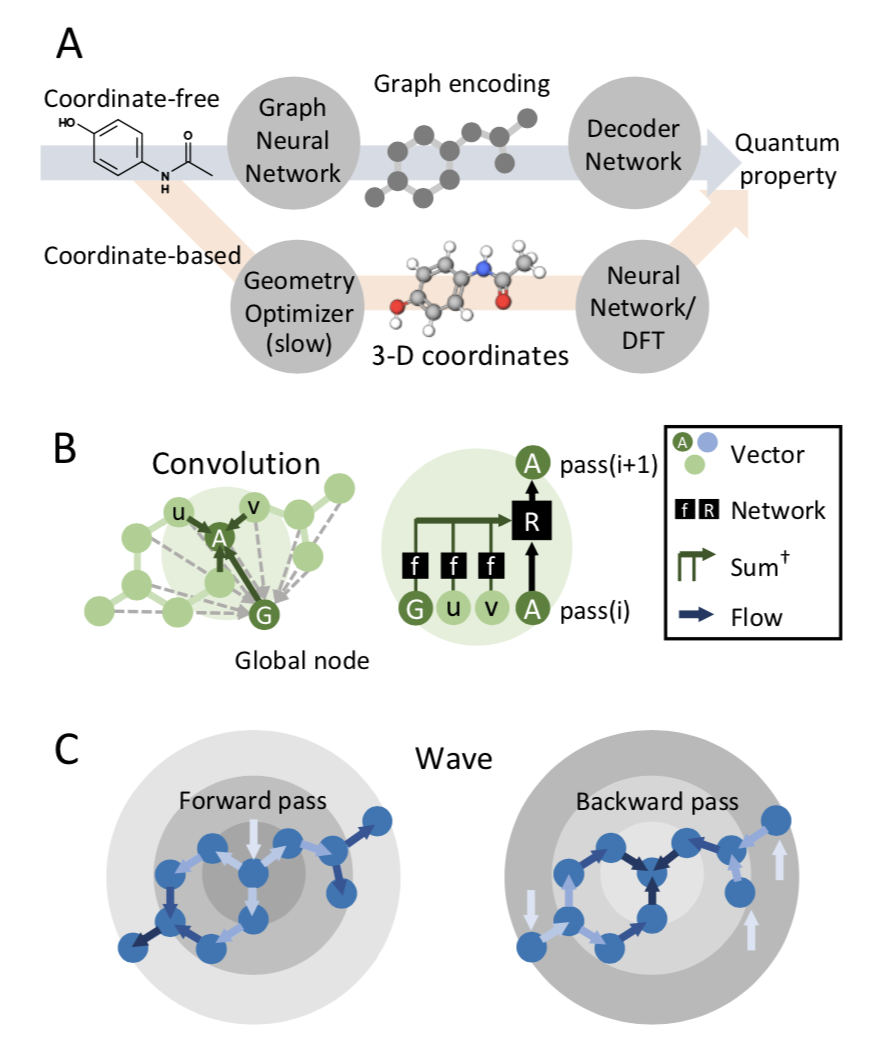 Computing quantum chemical properties of small molecules and polymers can provide in- sights valuable to physicists, chemists and biologists when designing new materials, catalysts, biological probes and drugs. Deep learning can compute quantum chemical properties accurately in a fraction of the time required by commonly used methods such as density functional theory (DFT). However, many of these deep learning architectures require energy minimized molecular geometries as input, which is also computationally expensive, and decreasing the reproducibility and throughput of these methods. In this study, we demonstrate that accurate quantum chemical computations can be performed without optimized geometries by operating in the coordinate-free domain using deep learning on graph encodings. We also find
that the choice of graph-encoding architecture substantially affects the performance of these methods. The Wave architecture outperforms graph convolution architectures, particularly on complex molecules. Furthermore, the structures of these graph encoding architectures provide an opportunity to probe an important, outstanding question in quantum mechanics: What types of quantum chemical properties can be represented by local-variable models? We find that Wave, a local-variable model, more accurately calculates quantum chemical properties. Graph convolutional architectures require global variables, and are not as effective as as Wave. We anticipate that coordinate-free, deep-learning models of quantum chemistry will become valuable tools in chemistry and biology, enabling researchers to rapidly screen chemical databases or identify new molecules using automated, de-novo design algorithms.
Computing quantum chemical properties of small molecules and polymers can provide in- sights valuable to physicists, chemists and biologists when designing new materials, catalysts, biological probes and drugs. Deep learning can compute quantum chemical properties accurately in a fraction of the time required by commonly used methods such as density functional theory (DFT). However, many of these deep learning architectures require energy minimized molecular geometries as input, which is also computationally expensive, and decreasing the reproducibility and throughput of these methods. In this study, we demonstrate that accurate quantum chemical computations can be performed without optimized geometries by operating in the coordinate-free domain using deep learning on graph encodings. We also find
that the choice of graph-encoding architecture substantially affects the performance of these methods. The Wave architecture outperforms graph convolution architectures, particularly on complex molecules. Furthermore, the structures of these graph encoding architectures provide an opportunity to probe an important, outstanding question in quantum mechanics: What types of quantum chemical properties can be represented by local-variable models? We find that Wave, a local-variable model, more accurately calculates quantum chemical properties. Graph convolutional architectures require global variables, and are not as effective as as Wave. We anticipate that coordinate-free, deep-learning models of quantum chemistry will become valuable tools in chemistry and biology, enabling researchers to rapidly screen chemical databases or identify new molecules using automated, de-novo design algorithms.