Effects of Charge Localization on the Orbital Energies of Bithiophene Clusters
Tamika A. Madison, Geoffrey R. Hutchison. “Effects of Charge Localization on the Orbital Energies of Bithiophene Clusters.” J. Phys. Chem. C 2011 115(35) pp 17558–17563. Available Online.
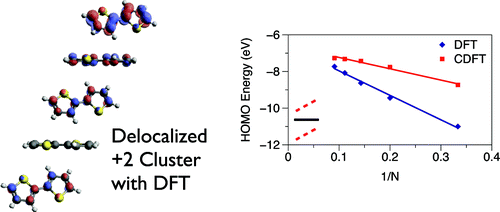 Standard and constrained density functional theory calculations were used to study the degree of charge localization in positively charged bithiophene clusters. While polarization effects due to the crystalline environment are known, many charge transport models in pi-conjugated organic materials assume a highly localized picture of carriers due to the strong electron-phonon interaction. These first- principles calculations show that that the positive charge delocalizes over at least 8 molecules in 1D herringbone stacks. For such 1D clusters, positive charge is more likely to localize on “tilted€” molecules than “parallel”€ molecules due to polarization effects. For 2D clusters, while polarization effects cancel due to symmetry, positive charge is shown to affect molecular sites anisotropically. Differences in computed HOMO energies between the localized and delocalized charges are ~1-2 eV, about the same as the difference in energy computed between a singly-charged and doubly-charged stack. These results suggest that models for charge transport in organic semiconductors should be revised to account for significant charge delocalization and intermolecular interactions such as polarization.
Standard and constrained density functional theory calculations were used to study the degree of charge localization in positively charged bithiophene clusters. While polarization effects due to the crystalline environment are known, many charge transport models in pi-conjugated organic materials assume a highly localized picture of carriers due to the strong electron-phonon interaction. These first- principles calculations show that that the positive charge delocalizes over at least 8 molecules in 1D herringbone stacks. For such 1D clusters, positive charge is more likely to localize on “tilted€” molecules than “parallel”€ molecules due to polarization effects. For 2D clusters, while polarization effects cancel due to symmetry, positive charge is shown to affect molecular sites anisotropically. Differences in computed HOMO energies between the localized and delocalized charges are ~1-2 eV, about the same as the difference in energy computed between a singly-charged and doubly-charged stack. These results suggest that models for charge transport in organic semiconductors should be revised to account for significant charge delocalization and intermolecular interactions such as polarization.